France
France Australia
Australia
30,000 Americans have Huntington’s disease (HD).1 It’s an inherited disorder that affects both sexes and all races. Within families, multiple generations may have the disease. But what causes HD? What are the symptoms? Learn more about Huntington’s disease.
- What is Huntington’s disease?
- What causes Huntington’s disease?
- What are the stages of Huntington’s disease?
- What are the symptoms of Huntington’s disease?
- HD Genetic Testing and Prevention
- Treatment for Huntington’s disease
- Common Difficulties as Huntington’s Disease Progresses
What is Huntington’s disease?
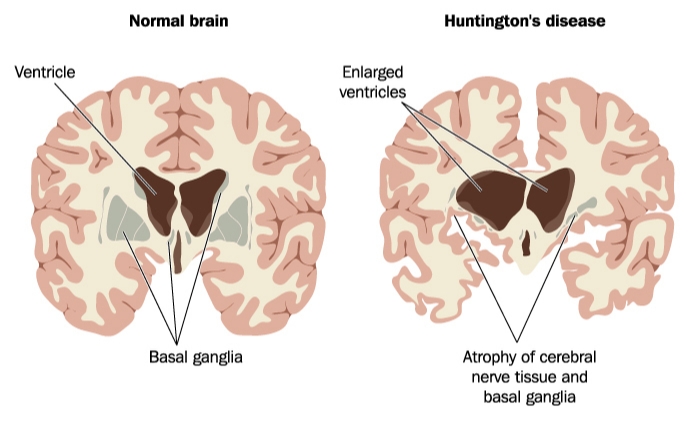
Huntington’s disease (HD) is an inherited disease that causes a progressive breakdown in the nerve cells of the brain. This disorder causes uncontrolled motor movements, the loss of thinking abilities (cognition), and emotional problems.
What causes Huntington’s disease?
It’s caused by a gene mutation.
Huntington’s disease is caused by a mutation in the HTT gene. HTT provides instructions for making the protein called huntingtin. The protein’s purpose isn’t completely known, but it appears to play an important role in the nerve cells of the brain.
Four bases make up the building blocks of DNA adenine (A), cytosine (C), guanine (G), and thymine (T). They are combined in various patterns to create DNA. The HTT mutation impacts a DNA segment called CAG. CAG typically repeats 10-35 times within the gene. In Huntington’s disease, the CAG segment is repeated 36-110+ times. People with 36-39 CAG repeats may or may not develop symptoms, while people with more than 40 repeats almost always develop the disorder.2
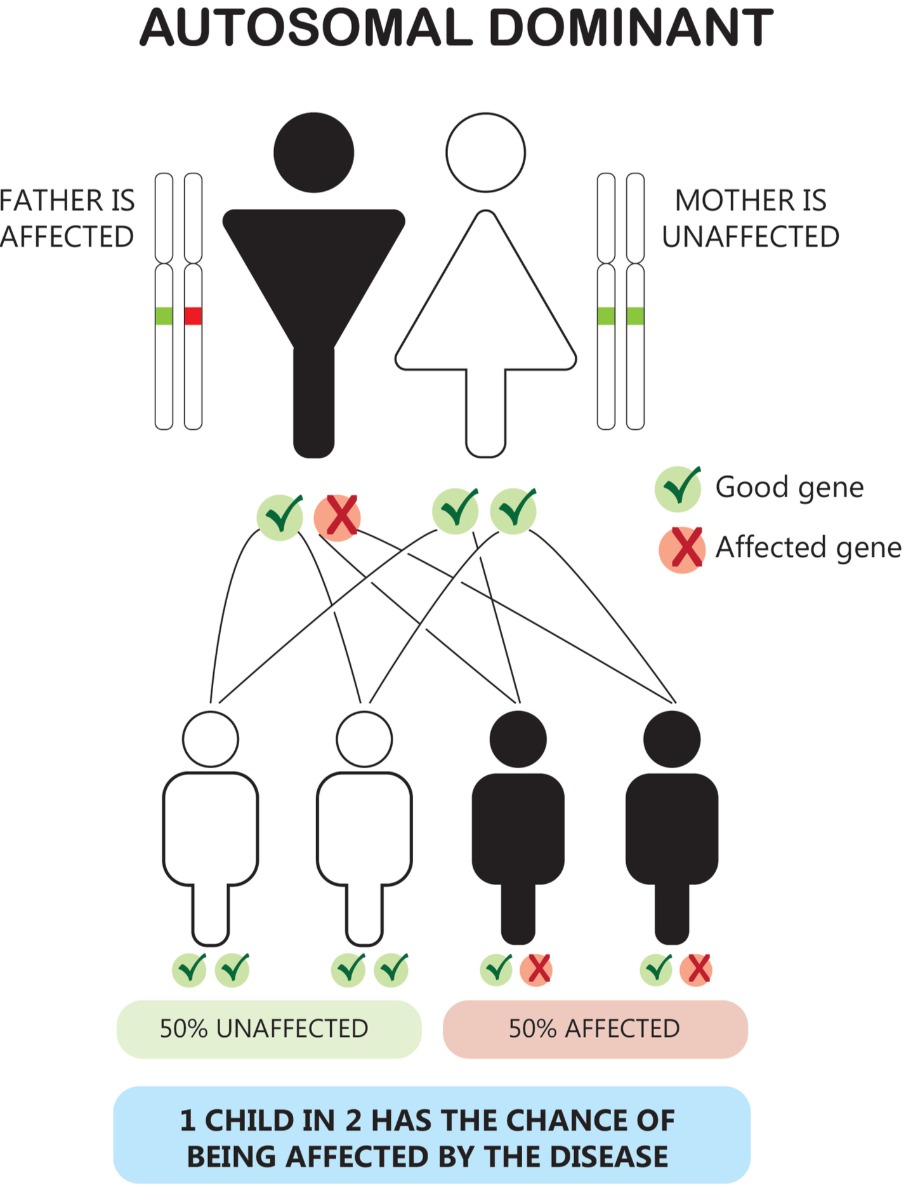
The gene mutation is typically inherited.
Huntington’s disease is typically inherited in an autosomal dominant pattern. This means that an affected person only needs to inherit one copy of the altered gene from one affected parent to have the disorder.
If one parent has the gene for Huntington’s disease and one parent does not have the gene, their children will have a 50% chance of inheriting the gene that causes HD.3
In 1-3% of people with HD, no family history of the disorder is ever identified.3
An increase in the CAG repeats can lead to an earlier onset.
As the altered HTT gene is passed from one generation to the next, the size of the CAG repeat often increases. This larger number of repeats often leads to an earlier onset of symptoms.
- 27-35 CAG repeats
- People with this number of CAG repeats do not develop Huntington’s disease, but they are at risk of having children with the disorder.2
- 40-50 CAG repeats
- Adult onset Huntington’s disease2
- 60+ CAG repeats
- Juvenile Huntington’s disease, about 10% of people with HD2
What are the stages of Huntington’s disease?
Huntington’s disease is typically broken into three stages. From disease emergence to death it is often 10-30 years, but disease progression including how long each stage lasts varies based on the individual.5
- Early stage
- During this stage, individuals are largely independent and can continue to work, drive, handle money, and live independently.
- Middle stage
- In this mid-stage, individuals with HD may no longer be able to work, drive, manage their finances, or perform household chores. However, they can eat, dress, and attend to their personal hygiene with assistance.
- Late stage
- In late stage HD, individuals require aid in all activities of daily living. They are typically non-verbal and confined to their bed, but seem to retain some comprehension.
What are the symptoms of Huntington’s disease?
The symptoms of Huntington’s disease vary based on the stage of HD that the individual is in. Symptoms typically begin when the person is in their 30s or 40s, but people with early onset HD develop symptoms in their 20s.
- Early stage symptoms
- Minor involuntary movements (chorea)
- Minor loss of coordination
- Difficulty thinking through complex problems
- Possible psychiatric symptoms, including depression
- Middle stage symptoms
- Prominent chorea
- Difficulty with voluntary motor coordination
- Problems swallowing
- Problems with balancing and falls
- Weight loss
- Difficulty problem solving due to an inability to organize, sequence, or prioritize information
- Possible psychiatric symptoms, including irritability or disinhibition
- Late stage symptoms
- Severe chorea may occur, but is often replaced by
- Rigidity
- Dystonia: muscle contractions that cause the body part to twist involuntarily
- Bradykinesia: unusually slow movements
- Speech impairment - often in this stage the individual is non-verbal
- Psychiatric symptoms may occur, but are harder to recognize due to communication difficulties
- Severe chorea may occur, but is often replaced by
HD Genetic Testing and Prevention
Genetic Testing
If you have symptoms that strongly suggest Huntington’s disease, a genetic test may be done to confirm the diagnosis. It may also be helpful in the determination of diagnosis if there’s no known family history of HD.
If you have a family history of HD, but do not have symptoms, you can get a predictive genetic test. The test cannot tell you when you will develop symptoms or what symptoms will occur first. This is a personal decision and testing is not done until after consultation with a genetic counselor.
Some people choose not to have the test because they would prefer not to know and do not see a benefit since there is not currently a cure. Other people want to be tested because they find the uncertainty stressful or want to know before having children.
Prevention: Prenatal Planning
If one parent has the Huntington’s disease gene, their child has a 50% chance of inheriting the disorder.3 However, if the family wants to have a child without the HD gene, there are a few options.
Pre-genetic diagnostic testing (PGD) and In Vitro Fertilization (IVF) can be used to ensure the implanted fertilized egg does not have the abnormal gene.4 This can be done without informing the at-risk parent of whether or not they have the HD gene, if they would prefer not to know.
If the woman is already pregnant, the fetus can be tested for the HD gene via chorionic villus biopsy at 10-11 weeks or amniocentesis at 14-18 weeks.4
Treatment for Huntington’s disease
There is currently no cure for Huntington’s disease. No treatment can alter the course of the disease, but there are some medications that can improve certain symptoms. There are also interventions that can help a person cope with their changing abilities.
Medications can help control motor movements and treat psychiatric disorders. Speech therapy can help with talking, eating, and swallowing. Physical therapy exercises may be able to enhance strength, flexibility, balance and coordination. Physical therapists can also provide instruction on using mobility aids. Occupational therapists can advise on assistive tools to use to improve eating, drinking, bathing, and dressing skills.
Talk to your doctor about possible treatment and therapy options for Huntington’s disease.
Common Difficulties as Huntington’s Disease Progresses
As Huntington’s disease progresses, eating, bathing, dressing, mobility, and other everyday tasks can become difficult. These articles share tips and products that can help you continue to live safely and independently.
- The Best Dining Aids for People with Huntington’s Disease
- 8 Bathing & Dressing Aids for Huntington’s Disease
- Huntington’s Disease: Managing Contractures
- Huntington’s Disease: Mobility Aids for Every Stage
- Helpful Transfer Aids for People with Huntington's Disease
- Fall Prevention for People With Huntington's Disease
References
- Huntington’s Disease Society of America. (n.d.). Who Is At Risk. Huntington’s Disease Society of America. Retrieved from https://bit.ly/2Zo8aIL
- Genetics Home Reference. (2020). Huntington disease. NIH U.S. National Library of Medicine. Retrieved from https://bit.ly/2ZmO01X
- John Hopkins Medicine. (n.d.). Huntington’s Disease. John Hopkins Medicine. Retrieved from https://bit.ly/2XkBtcC
- Huntington’s Disease Society of America. (n.d.). Genetic Testing & Family Planning. Huntington’s Disease Society of America. Retrieved from https://bit.ly/2AQ5XvD
- Mayo Clinic Staff. (2020). Huntington’s disease. Mayo Clinic. Retrieved from https://mayocl.in/2LNq2F5
- Huntington’s Disease Society of America. (n.d.). Caregiver Guide for Mid to Late Stage Huntington’s Disease: For Long-Term Care Facilities and In-Home Care Agencies. Huntington’s Disease Society of America. Retrieved from https://bit.ly/3g5izzb
- Huntington’s Disease Society of America. (n.d.). Age of Onset. Huntington’s Disease Society of America. Retrieved from https://bit.ly/2yrhZL8
- Huntington’s Disease Society of America. (n.d.). Huntington’s Disease Stages. Huntington’s Disease Society of America. Retrieved from https://bit.ly/36omjHp
Medical Disclaimer: The information provided on this site, including text, graphics, images and other material, are for informational purposes only and are not intended to substitute for professional medical advice, diagnosis or treatment. Always seek the advice of your physician or other healthcare professional with any questions or concerns you may have regarding your condition.